INTRODUCTION
Malignant peripheral nerve sheath tumors (MPNSTs) are rare soft tissue sarcomas that occur in 8%–13% of patients with neurofibromatosis type 1 (NF1), a neurocutaneous condition characterized by skin discoloration. Meanwhile, in the general population, the incidence of MPNSTs has been estimated to be 0.001% [1,2]. NF1 is one of the most important risk factors for MPNSTs [3]. MPNSTs often present as mass-like lesions of the head, neck, body, and other anatomical areas, with pain associated with neural system involvement [3].
MPNSTs are often confused with neurofibromas or plexiform neurofibromas in patients with NF1. As a result, there may be a delay in diagnosis and proper treatment [4]. Wide radical resection with negative margins is therapeutically effective, especially when followed by adjuvant radiotherapy [5]. Herein, we report a case of a patient with MPNST that was accidentally identified after two surgical resections, the specimens from which were diagnosed as neurofibroma, in a known NF1 patient.
CASE REPORT
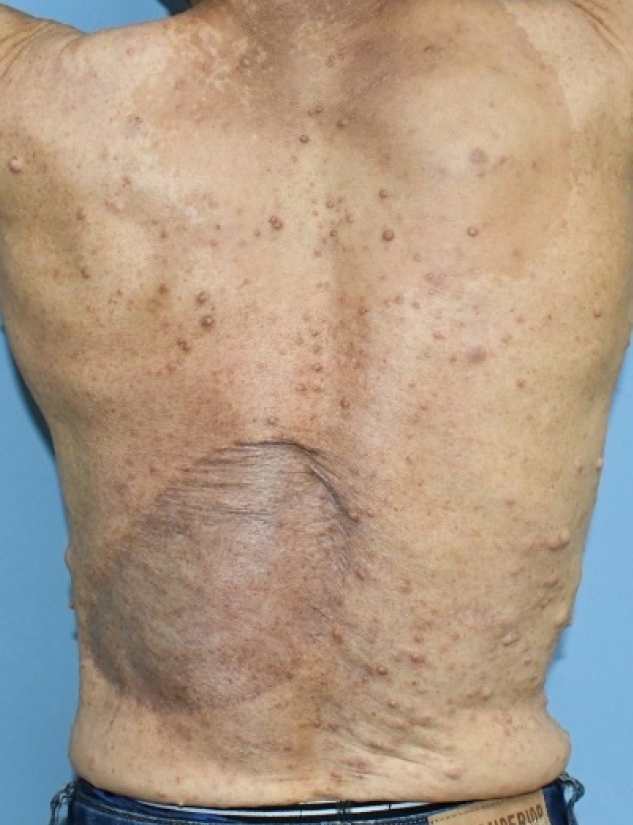
A 55-year-old man with known NF1 visited our hospital. The patient had a mass measuring 12 × 20 cm on his back and complained of discomfort in daily life. He had no history of radiation therapy. Physical examination revealed café-au-lait spots and multiple small masses across his entire body that were consistent with NF1; in addition, a large mass was visible on the left side of the back that was soft and tender upon palpation (Fig. 1). The patient had previously undergone surgery to remove the masses from the back on two occasions at the department of general surgery. The specimens from both operations were diagnosed as neurofibroma based on histopathological and immunohistochemical evaluations by a pathologist. An ultrasonographic examination showed a well-defined, hypoechoic mass in the subcutaneous layer without any evidence of muscle invasion (Fig. 2). An enhanced computed tomography (CT) scan revealed a heterogeneous, subcutaneous, globular mass lesion in the upper posterior abdominal wall and confirmed the diagnosis of neurofibroma (Fig. 3).
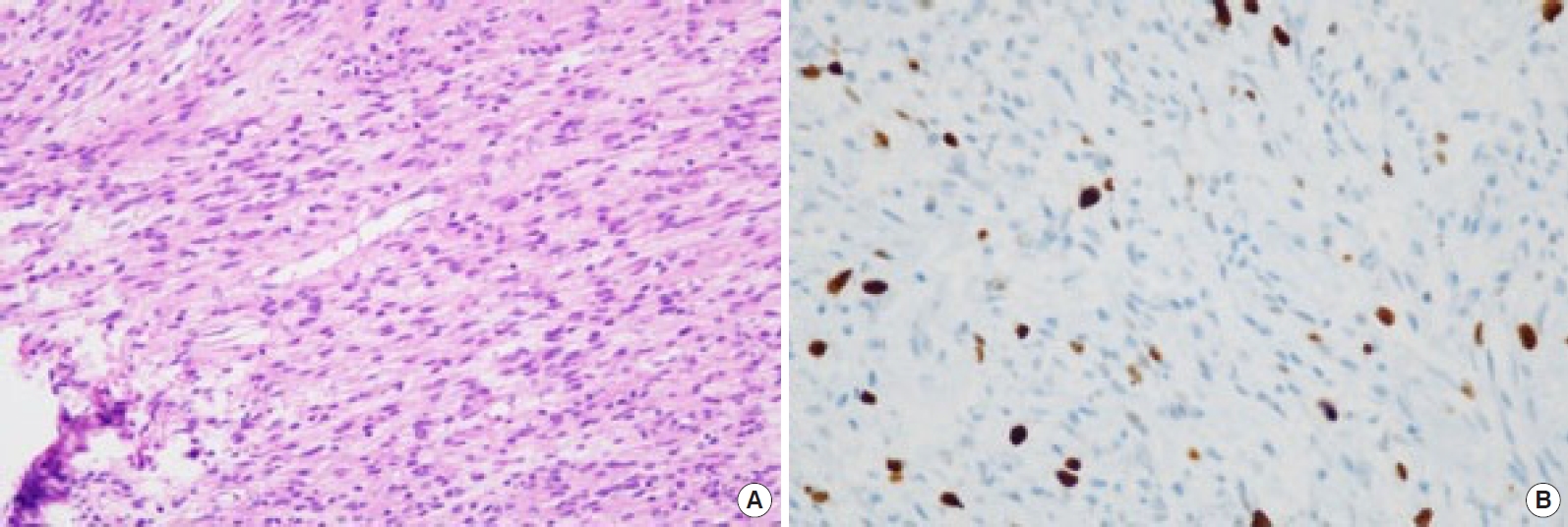
The radiological evaluation and histopathological examination of specimens from the previous two operations had confirmed the diagnosis of neurofibroma. Hence, with the patient under general anesthesia, we performed total excision of the mass and skin grafting. A mass measuring approximately 12 × 20 cm was excised from the patient’s back, after which a split-thickness skin graft was performed (Fig. 4). The resected specimen was sent for microscopic examination, and immunohistochemical staining was performed. A typical herringbone pattern and diffuse atypical mitoses were observed in the histopathological examination, and immunohistochemical staining revealed positive expression (20%) of the proliferation marker Ki-67 (MKI67) antigen (Fig. 5). The biopsies confirmed the diagnosis of MPNST associated with NF1. All resection margins were confirmed to be negative. In order to determine the extent of metastasis, the patient underwent positron emission tomography with fluorine-18-fluorodeoxyglucose. There was no significant hypermetabolic activity suggestive of metastasis. We advised the patient to undergo radiotherapy, which subsequently began 2 months after the operation. Daily adjuvant radiotherapy was administered at a dose of 180–200 cGy, amounting to a total dose of 50–55 Gy over a period of 6 weeks.
The patient was followed up in the outpatient department at 1, 6, and 18 months. The patient’s condition remained stable, and there were no postoperative complications at the operation site (Fig. 6).
DISCUSSION
MPNSTs are rare malignant soft tissue sarcomas that occur in the peripheral nerves, accounting for approximately 5%–10% of all soft tissue sarcomas [4,6]. Existing benign plexiform neurofibromas, a history of prior radiation treatment, large size, and mutation of the NF1 gene and surrounding genes are risk factors for the development of MPNSTs [7].
Early diagnosis of MPNSTs is important, and wide radical resection with negative margins is essential. However, distinguishing among MPNSTs, neurofibromas, and plexiform neurofibromas remains a clinical challenge. MPNSTs are often misdiagnosed in small samples such as those obtained by needle biopsy [8]. As a result, the diagnosis of MPNSTs is frequently delayed. Furthermore, the clinical features of MPNSTs, such as pain and neurological compromise, are similar to those of neurofibromas [2]. On ultrasonography, MPNSTs typically appear as well-defined, fusiform, hypo echoic masses [9]. CT can be used to determine the location and size of MPNSTs and to identify the presence of heterogeneous attenuation due to necrosis or hemorrhage. Ultrasonography and CT are rarely useful when differentiating between benign and malignant tumors [10]. Magnetic resonance imaging (MRI) may be useful for differentiating MPNSTs from neurofibromas. The presence of statistically significant MRI features is highly suspicious for malignancy. However, the diagnosis of MPNSTs should not rely solely on imaging; instead, all suspected cases should be biopsied [9,11].
Typically, radiological examinations of patients with MPNSTs reveal findings such as giant masses (> 5 cm), bony destruction, intratumoral lobulation, heterogeneous enhancement, perilesional edema, and local invasiveness [9,10,12]. If two or more of the aforementioned features are present, then an early diagnosis can be given [13]. Histologically, MPNSTs typically show a pattern of fasciculated growth of hyperchromatic spindle cells. Other common features include an elongated nucleus, perivascular hypercellularity, pronounced mitotic activity, and focal tumor necrosis [14]. Immunohistochemical studies using cell proliferation markers such as the MKI67 antigen have shown promise for predicting the prognosis of neoplasia. Moreover, the presence of the tumor protein p53, a tumor suppressor protein, has been reported in various soft tissues and neuronal neoplasms in which MKI67 was overexpressed [15].
In this report, we present a case where MPNST occurred following the diagnosis of neurofibroma, which was given based on biopsies performed after two separate operations. At first, we diagnosed the giant mass as a neurofibroma; however, it was diagnosed as MPNST following histopathological and immunohistological examinations of the resected specimen. Hence, we decided to perform adjuvant radiotherapy. Since MPNST is a life-threatening form of sarcoma, early diagnosis and wide radical excision are key to successful treatment. The possibility of malignant tumors should always be considered in NF1 patients presenting with very large masses.