INTRODUCTION
Juvenile xanthogranuloma (JXG) is the most common type of nonLangerhans cell histiocytosis in infancy and early childhood [1]. Classic JXG is often located on the skin of the head, neck, and upper trunk and is characterized by yellowish, asymptomatic, elevated skin nodules measuring 0.5 to 2.0 cm in diameter. In general, JXG often regresses over 3 to 6 years and is more predominant in boys than in girls. Most are already present at birth, and the prevalence of JXG may be underestimated since some lesions spontaneously regress.
JXG is classified into cutaneous and non-cutaneous types according to the characteristics of the lesion. Cutaneous JXG usually progresses through a benign course, followed by involution within months or years. Therefore, persistence of the disease into late childhood has been rarely reported [2]. Non-cutaneous JXG occurs in 5% of children and usually presents in the eye, lung, heart, gastrointestinal tract, central nervous system, adrenal gland, pituitary gland, bone, bone marrow, or kidney, but the uveal tract of the eye is the most common site [3].
Most of the previously reported cases were cutaneous and noncutaneous JXG in children. We present a rare case of JXG in the axillary area that suddenly developed in a 36-year-old male patient.
CASE REPORT
A 36-year-old male patient was admitted to the outpatient clinic with a swollen red nodule on the axilla that had been quickly growing over the previous 2 months. He complained of an occasional itching sense in the axilla. He stated that he had not had any mass lesions since childhood, and did not take any medications for this lesion. The patient did not have any other known underlying diseases, had no unusual environmental exposures, and was not a smoker. His initial laboratory studies were within normal limits.
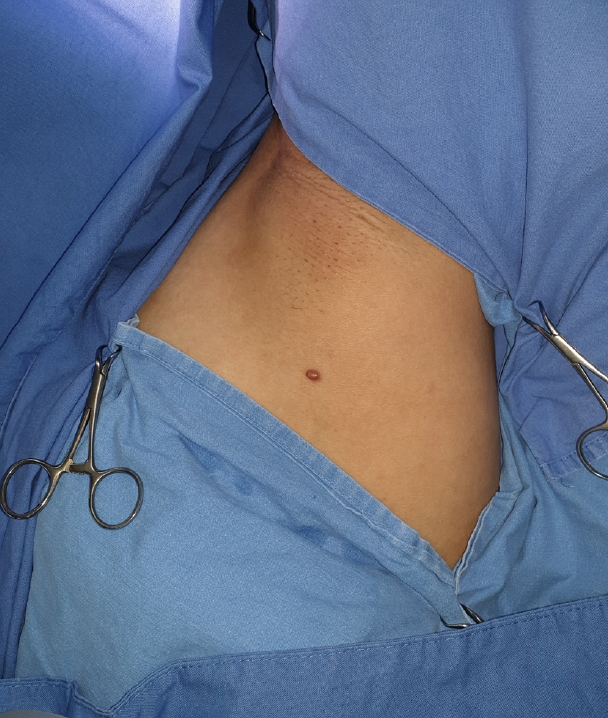
On physical examination, the lesion appeared as a non-tender, mass-like lesion that measured 1.5 cm in diameter on the axilla (Fig. 1). Although we recommended a punch biopsy and additional imaging studies for the differential diagnosis, the patient requested a further evaluation if a malignancy was confirmed after surgical treatment.
We planned surgical resection of the tumor under local anesthesia. We performed en-bloc excision with a safety margin of 0.5 cm from the mass. The mass was a well-differentiated red nodule, and no fluid was found in the cavity (Fig. 2). We performed a frozen biopsy during the operation, and there was no suspicion of malignancy within the safety margin.
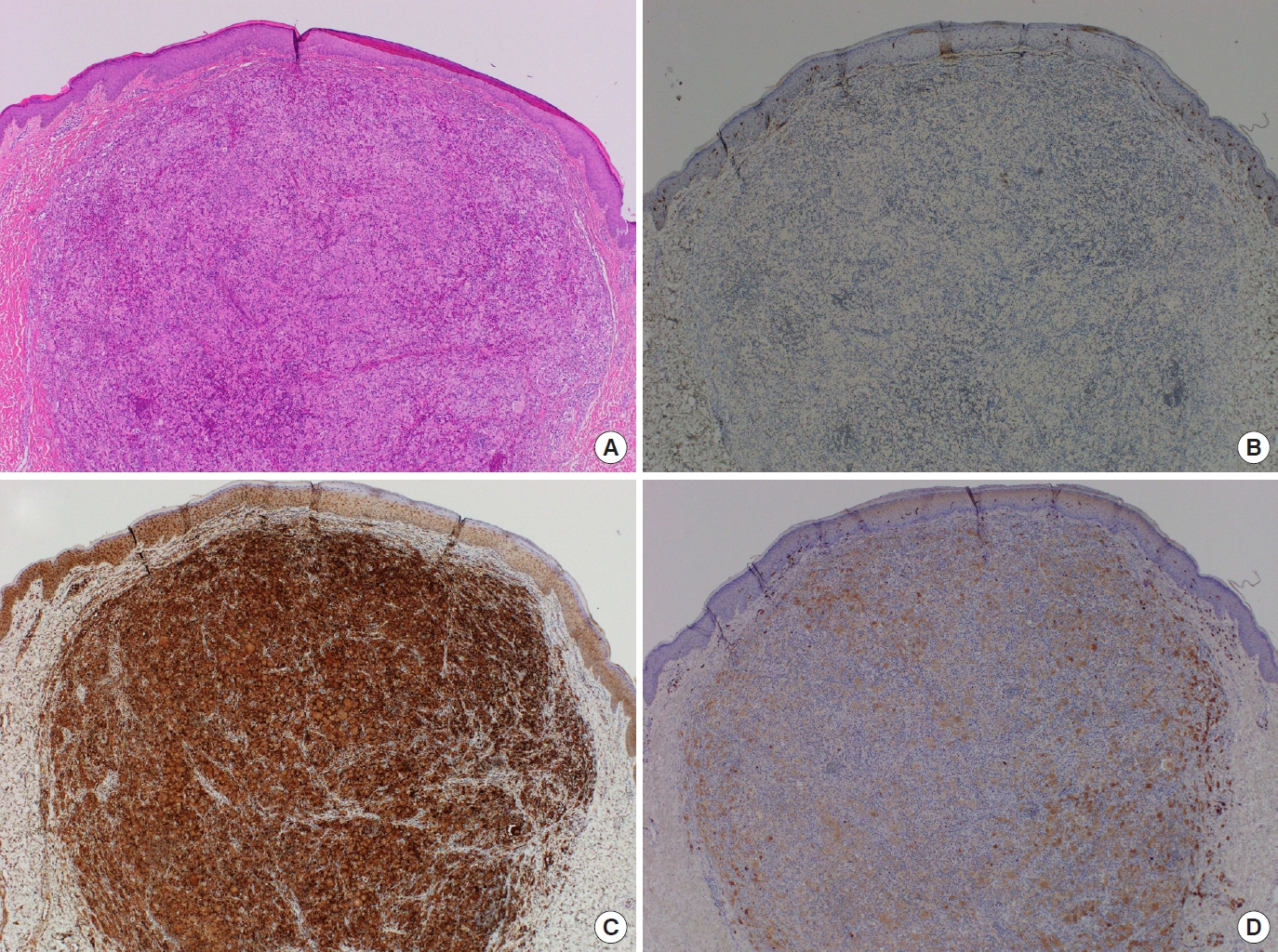
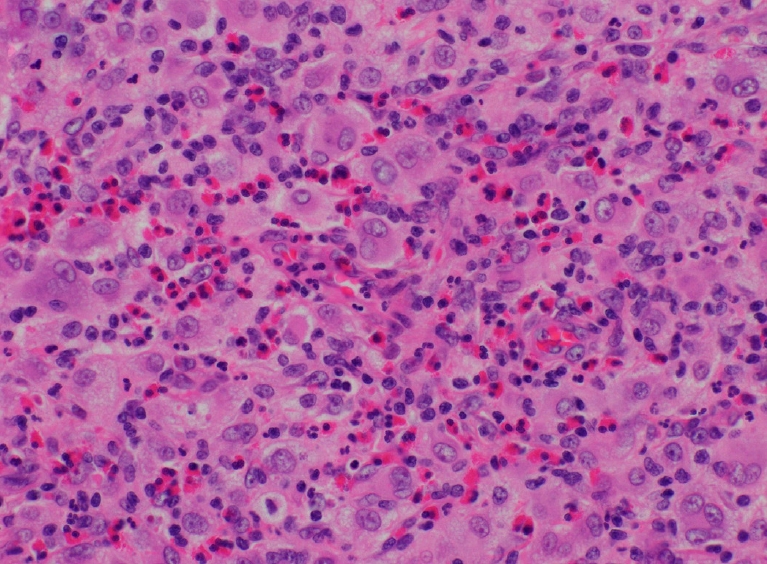
Histopathological examination revealed JXG with dermal infiltration of foamy histiocytes and scattered giant cells. In an immunohistochemical examination, the specimen stained positive for CD68 and S-100 and negative for CD1a, which is a reasonable finding for JXG (Figs. 3, 4).
At a 3-month postoperative follow-up, the patient had completely recovered with no complications (Fig. 5). He reported no limitations in movement of the axillary area when exercising. The patient will continue to be observed through outpatient follow-up.
DISCUSSION
JXG is a rare skin disease arising from dermal dendrocytes, which are non-Langerhans cell histiocytes, and is a kind of dendritic cellrelated histiocytic disorder. In particular, JXG is a granulomatous inflammatory lesion commonly seen in childhood. In 1997, the World Health Organization declared that both JXG and Langerhans cell histiocytosis are on the spectrum of dendritic cell-related diseases [4].
JXG characteristically appears as single or multiple cutaneous, elevated, yellowish-brown, painless lesions at a mean age of 3 years. Our patient had a solitary red nodule with tenderness on his axilla. Although little is known about the pathogenesis of JXG, several recent genetic studies have shown that complex genomic factors may be involved [5].
In the literature, most cases of JXG were reported in childhood and infancy, and the characteristics of their disease were also similar. Hoeger and Patrizi reported cutaneous JXG lesions that involved the face, eyelids, and the inguinal and genital areas [6,7].
Shani-Adir et al. [8] reported a combined case in a 2-year-old boy who was diagnosed with Langerhans cell histiocytosis that involved the right tibia and non-Langerhans cell histiocytosis of the face, neck, and upper trunk.
A congenital type of JXG has also been reported. Chubak et al. [9] reported a 2-week-old boy who had congenital eyelid cutaneous JXG that was treated with an intralesional steroid without surgical excision.
Oza et al. [10] identified 31 cases of congenital JXG that involved the skin or presented with systemic involvement. Patients with multiple cutaneous lesions must undergo a further systemic evaluation. Parents who find lesions only on the skin usually bring their infants to the hospital, but the medical team must conduct a systemic evaluation.
Chauhan et al. [11] argued for the importance of fine needle aspiration (FNA) cytologic biopsy to determine the need for surgical treatment. In our case, we recommended FNA biopsy to the patient, but he requested complete excision of the lesion.
In addition, an immunohistochemical study is essential in the differential diagnosis of JXG and Langerhans cell histiocytosis. In immunohistochemical studies, JXG is positive for macrophage markers (CD 68, CD163, vimentin, and anti-CD4) and occasionally negative for S-100, CD1a, and CD207 [12].
In general, JXG has a good prognosis of disease progression with spontaneous remission, and relapsing lesions are rare. If a patient has an isolated, single, cutaneous lesion, then an en-bloc excision is recommended for complete cure [13].
Clinical observation is appropriate in children with spontaneously regressing lesions. However, cases of multiple lesions with systemic disease (visceral organs, central nervous system, and so on) require a diagnostic work-up to assess the possible presence of other combined diseases.
Nolte et al. [14] reported a large intracardiac JXG in a 50-yearold woman who had previously undergone mitral valve surgery. In addition, several cases have been reported of JXG involving the multiple sites of the iris, intracranial region, and neck occurring in adults in their 20s and 30s, showing that JXG may be prevalent in adulthood [3,15].
Therefore, JXG can arise from an older lesion, and adults still need clinical follow-up for JXG.
In summary, JXG is usually a self-limited, benign histiocytosis, and skin lesions usually resolve spontaneously without symptoms. Cases in adulthood, however, require systemic evaluation and proper treatment. Occasionally, complete excision is performed for the differential diagnosis, and some patients require close observation. The usual differential diagnosis of JXG in children includes Langerhans cell histiocytosis, other xanthomatous lesions, mastocytoma, Spitz nevus, and dermatofibroma, while in older children and adults, it includes xanthoma disseminatum and eruptive xanthoma. Adult xanthogranuloma is more solitary and larger than JXG, and is not characterized by the possibility of associated systemic diseases and spontaneous regression.
As cutaneous JXG lesions are self-healing, treatment is only necessary for diagnostic or cosmetic lesions. Spontaneous regression occurs in approximately 1 to 5 years. In our case, the mass grew rapidly over a 2-month period, and it was necessary to differentiate it from a malignancy, such as dermatofibrosarcoma. To rule out the possibility of malignancy, we performed a wide excision, but this will often cause a poor aesthetic result.
In conclusion, clinicians who diagnose JXG in an adult must recommend a further systemic evaluation and FNA biopsy before deciding upon the proper treatment. Appropriate treatment and patient management should be planned based on the preoperative findings.
Because not much is known about the pathogenesis of JXG, future studies should explore therapeutic targets as research on its pathogenesis progresses.